Python 自动化提取基因 CDS
环境准备及背景介绍
目录结构:

2
Python 实现
BaimoTools.py
1#!/usr/bin/env python2# -*- coding: utf-8-*-3# @Author : Baimoc 4# @Email : baimoc@163.com 5# @Time :2021/3/1714:286# @File : BaimoTools 7import os 8import time 910from Bio import SeqIO, SeqFeature 111213class BaimoTools():14 def __init__(self, gb_file, fasta_file):15 self.complete_fasta =""16 self.fasta_file = fasta_file 17 self.gb_file = gb_file 18 self.feature = None 19 self.record = None 2021 def format_val(self, object=None):22""" 23 格式化对象值为字符串 24 :param object: 对象或对象键值 25 :return: 26 """27 key =""28 # 判断参数是否为字符 29ifisinstance(object, str):30 obj = self.feature.qualifiers 31 key = object 32else:33 obj = object 34 # 为字符,提取 feature.qualifiers 对象关键字 35if key !="" and not obj.get(key):36return""37 elif key =="":38 val = obj 39else:40 val = obj[key]41 # 转换为字符串 42if not len(val):43 val =""44 elif len(val)==1:45 val = val[0]46else:47ifisinstance(val, SeqFeature.CompoundLocation) or isinstance(val, SeqFeature.FeatureLocation):48 val =str(val)49else:50 val =" | ".join(val)51return val 5253 def extract_cds(self, cds):54""" 55 获取 CDS 的 Fasta 序列 56 :param cds: 获取指定基因的 CDS 区域,如果为空,则获取全部 57 """58 records =list(SeqIO.parse(self.gb_file,"genbank"))59for record in records:60print(f"{record.id}")61for feature in record.features:62 # 提取 CDS 信息 63try:64if feature.type =="CDS":65 self.feature = feature 66 self.record = record 67 cds_gene = self.format_val('gene')68if cds =="":69 self.complete_fasta += self.format_fasta()70 elif isinstance(cds, str) and cds_gene == cds:71 self.complete_fasta += self.format_fasta()72 elif isinstance(cds, list) and cds_gene in cds:73 self.complete_fasta += self.format_fasta()74 except:75print(f"{record.id}: No CDS features")76 self.write_file()7778 def write_file(self):79""" 80 写入文件 81 """82withopen(self.fasta_file,"w")as f_obj:83 f_obj.writelines(self.complete_fasta)8485 def format_fasta(self, num=0):86""" 87 整理 Fasta 格式 88 :param num: 每行字符数,超出则换行 89 :return: Fasta 文本 90 """91 cds_gene = self.format_val('gene')92 cds_location = self.format_val(self.feature.location)93 cds_product = self.format_val('product')94 cds_protein_id = self.format_val('protein_id')95 cds_translation = self.format_val('translation')96 complete_ana = f">{self.record.id} | {cds_gene} | {cds_product} | {cds_protein_id} | {str(cds_location)}\n"97 format_seq =""98if num:99for i, char inenumerate(cds_translation):100 format_seq += char101 if(i +1)% num ==0:102 format_seq +="\n"103else:104 format_seq = cds_translation105 return complete_ana + format_seq +"\n"
3
使用示例
1
数据介绍
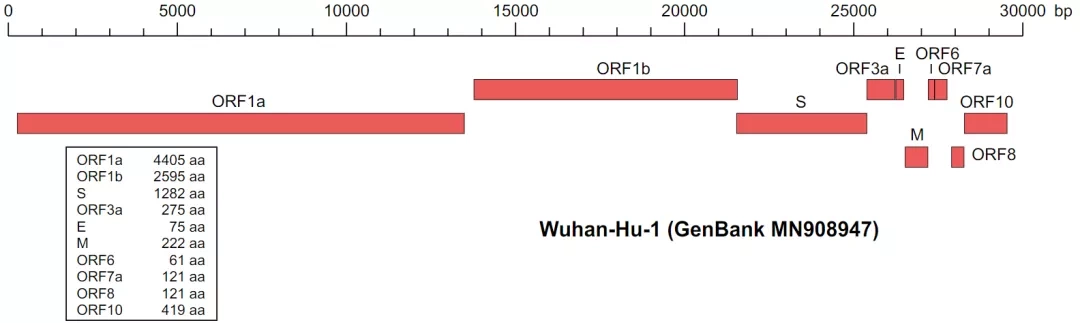
示例数据为新冠病毒的基因组 genbank 文件,文件中包含:
两个基因组:LC553263.1 和 LC553262.1
一个基因组会有多个基因,下面是它的基因组结构:
2
提取单个基因CDS
main.py
1from BaimoTools import BaimoTools23gb_file = f"res/genbank/SARS-CoV-2.gb"4fasta_file = f"out/output_s.fasta"5baimoTools =BaimoTools(gb_file, fasta_file)6# baimoTools.extract_cds('S')输出文件 output_s.fasta,分别提取到两个基因组的 S 基因 CDS 区域:
3
提取多个基因CDS
main.py
1from BaimoTools import BaimoTools23gb_file = f"res/genbank/SARS-CoV-2.gb"4fasta_file = f"out/output_s_m_orf10.fasta"5baimoTools =BaimoTools(gb_file, fasta_file)6baimoTools.extract_cds(['S','M','ORF10'])输出文件 output_s_m_orf10.fasta,分别提取到两个基因组的 S,M,ORF10 基因 CDS 区域::
4
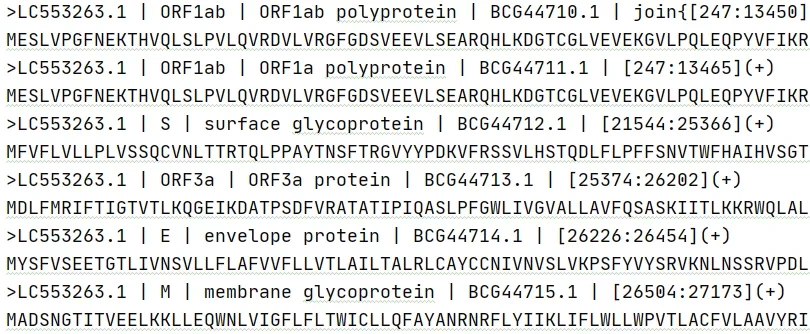
提取全部基因CDS
main.py
1from BaimoTools import BaimoTools23gb_file = f"res/genbank/SARS-CoV-2.gb"4fasta_file = f"out/output_s.fasta"5baimoTools =BaimoTools(gb_file, fasta_file)6# baimoTools.extract_cds("")输出文件 output_all.fasta,分别提取到两个基因组的全部基因 CDS 区域:
下一步更新其他基因特征提取,及格式转换功能。
以上是 Python 自动化提取基因 CDS 的全部内容, 来源链接: utcz.com/p/217218.html